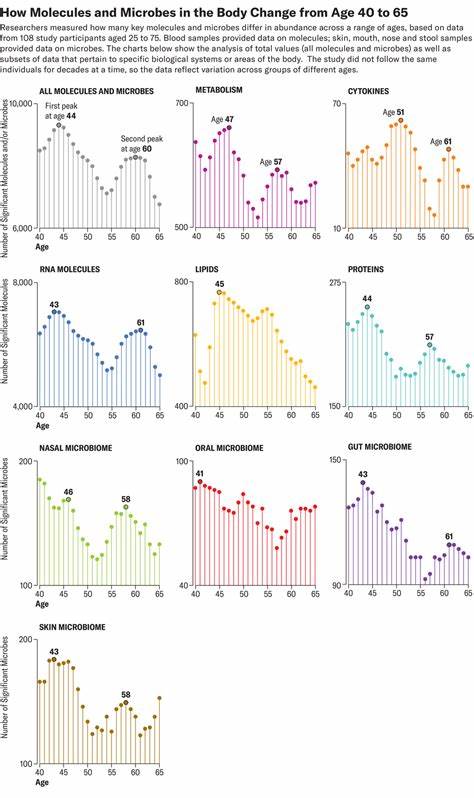
Определение видов и классификация биологических организмов всегда были ключевыми задачами биологии. Традиционные методы, основанные на морфологических признаках, часто сталкивались с трудностями, особенно при работе с родственными видами или фрагментами организмов. В последние десятилетия молекулярные методы, в частности ДНК-баркодирование, стали революционным инструментом для идентификации видов, способствуя развитию биоразнообразия, экологии, палеонтологии и многих прикладных наук. Несмотря на свою эффективность, классические методы ДНК-баркодирования имеют ограничения, связанные с выбором специфических генов, недостаточной универсальностью и низкой разрешающей способностью при определении близкородственных организмов. В ответ на эти вызовы появился инновационный подход — универсальная ДНК-подпись, построенная на основе использования данных низкого покрытия геномного сканирования и мощных алгоритмов машинного обучения.
Эта технология способна превратить разрозненные небольшие фрагменты генетической информации в уникальное графическое представление, отражающее целостный геномный «отпечаток» вида, что значительно расширяет возможности молекулярной идентификации и делает ее применимой для всего дерева жизни. Проблемы традиционного ДНК-баркодирования связаны с необходимостью подбора универсальных генетических маркеров. Чаще всего используются единичные гены или их фрагменты, например, COI у животных или matK и rbcL у растений. Однако такие локусы зачастую недостаточно информативны для отличия близких видов, а также не подходят для всех таксонов. Процесс амплификации и секвенирования может осложняться при низком качестве или фрагментированности исходной ДНК, присутствии паралогов и псевдогенов.
Кроме того, выделение и секвенирование конкретных баркодных локусов требует времени и ресурсов. В результате потребовались альтернативные методы, которые бы были универсальнее, не требовали точного знания генома и сохраняли высокую точность при минимальном объеме данных. Современный подход, представленный исследователями под названием varKoding, предлагает решение этих задач. Метод основан на использовании технологии «геномного сканирования» (genome skimming), при которой получают низкокачественные, но охватывающие весь геном данные с небольшим общим объемом секвенирования. Вместо поиска и анализа отдельных генов новая методика превращает считывания ДНК в двумерные изображения, представляющие частотный профиль коротких фрагментов нуклеотидов — к-миров.
Это визуальное отображение отражает разнообразные геномные особенности, включая состав нуклеотидов, повторяющиеся последовательности и другие характеристики, а не только классическое сравнение последовательностей. На подготовленных таких изображениях обучаются глубокие нейросети, способные распознавать и классифицировать виды с высокой точностью. В основе реализации varKoding лежит оригинальный способ преобразования частот к-миров в пиксели изображения, что позволяет учитывать вариации геномных признаков сложным и информативным образом. За счет применения трансформеров, в частности Vision Transformer (ViT), и оптимизированных хаотических игровых представлений обработка изображений выходит на качественно новый уровень. Такой подход показывает точность идентификации выше 91% даже при минимальном объеме данных, что превосходит существующие альтернативные методы.
Практическая реализация varKoding была проверена на разнообразных группах организмов, в том числе на представителях цветковых растений (Malpighiales), жуков, грибов и бактерий. Результаты демонстрируют стабильность и универсальность метода не только для видов, но и для родовых и семейных таксонов. Особое значение технология имеет для работы с образцами низкого качества, например, из гербариев или ископаемых останков, где традиционные методы бессильны из-за деградации ДНК и необходимости амплификации. Кроме того, varKoding обладает выдающейся масштабируемостью, позволяя обрабатывать сотни тысяч и миллионы образцов, используя ограниченный объем данных, который можно быстро получить с помощью портативного секвенатора Nanopore. Нейросетевые модели обучаются эффективно, имеют фиксированный размер, а сама технология не требует затратных этапов сборки генома или вычислений на основе парных сравнений каждого образца с каждым другим.
Это открывает возможности широкомасштабного мониторинга биоразнообразия и судебно-экологических исследований в полевых условиях. Масштабные испытания, проведенные с использованием данных из Национального Центра Биотехнологической Информации (NCBI) и глобальных воздушно-почвенных метагеномов, показывают высокую устойчивость метода к различиям в методах секвенирования, а также к разнородным сценариям применения, например, для определения налогономической принадлежности или географического происхождения образцов. Концептуально varKoding демонстрирует, что интеграция биоинформатики и современных методов машинного обучения способна преодолеть фундаментальные ограничения классической молекулярной таксономии. Представление геномных данных в виде изображений и применение алгоритмов компьютерного зрения открывают новые горизонты для исследования эволюции и биоразнообразия, предоставляя биологам мощный инструмент для быстрой и точной идентификации даже самых сложных и малоизученных таксонов всего древа жизни. Использование varKoding обещает не только улучшить научное понимание природы, но и содействовать прикладным задачам, таким как борьба с браконьерством, контроль качества медицинских растений, идентификация сельскохозяйственных вредителей и анализ экологических изменений на планете.




![Study shows link between CT scans and the risk of developing cancer [video]](/images/C7042551-39E7-4FDA-99B1-669C7A3799DA)